Riechstörungen manifestieren sich in verschiedenen Ausprägungen, wobei zwischen einem vollständigen Verlust oder Fehlen der Riechfunktion (Anosmie) und einer quantitativen Reduktion derselben (Hyposmie) unterschieden wird. Des Weiteren sind Riechstörungen zu verzeichnen, die mit einer abnormen Wahrnehmung von Gerüchen (Parosmie) einhergehen. Die Auswirkungen einer beeinträchtigten Riechfunktion erstrecken sich auf diverse Aspekte des täglichen Lebens. Insbesondere sind potenzielle Beeinträchtigungen in Bereichen wie häusliche Sicherheit (z.B. aufgrund mangelnder Wahrnehmung von Brandgefahren oder Rauch), Ernährung (z.B. durch den Konsum verderblicher Lebensmittel) und soziale Interaktionen (aufgrund der fehlenden Wahrnehmung des eigenen Körpergeruchs) zu berücksichtigen.
Ursachen und Häufigkeit von Riechstörungen im Kindes- und Jugendalter
Im Kontext der Riechstörungen im Kindes- und Jugendalter lassen sich zwei grundlegende Kategorien identifizieren: angeborene und erworbene Formen. Bei kongenitalen Riechstörungen werden die Kinder bereits ohne Geruchssinn geboren. Diese angeborenen Riechstörungen können sowohl isoliert als auch in Verbindung mit bestimmten Syndromen, wie dem Kallmann-Syndrom oder dem CHARGE-Syndrom, auftreten. Interessanterweise manifestieren sich angeborene Riechstörungen zwar von Geburt an, werden jedoch durchschnittlich erst im Alter von etwa 10 Jahren wahrgenommen. Die Diagnose „kongenitale Anosmie“ wird oft erst nach vielen Jahren gestellt, was für die Betroffenen eine langwierige und belastende Zeit darstellen kann, begleitet von zahlreichen Arztbesuchen.
Erworbene Riechstörungen können im Kindes- und Jugendalter durch verschiedene Ursachen entstehen, wie beispielsweise Schädel-Hirn-Trauma, vergrößerte Rachenmandeln (Adenoid Hypertrophie) oder postinfektiöse Komplikationen.
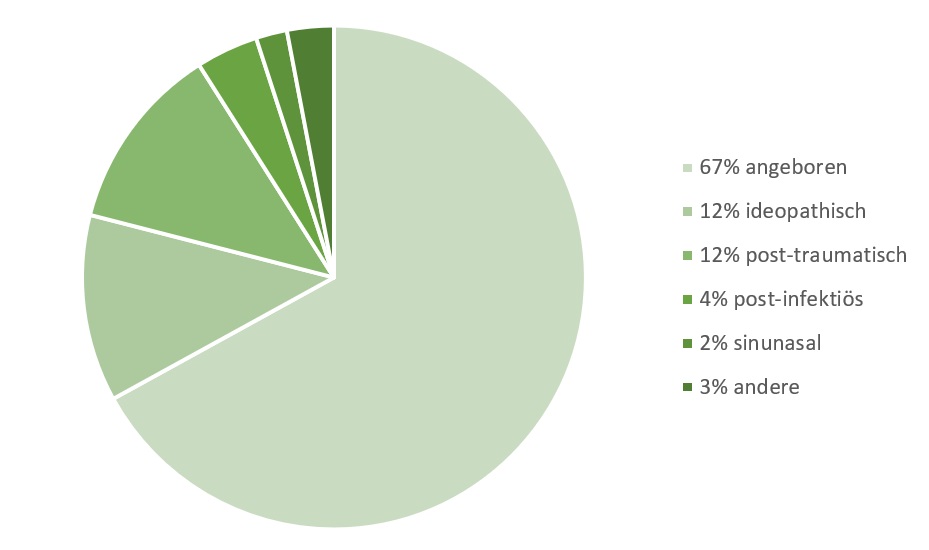
Die Mehrheit der Kinder und Jugendlichen, die aufgrund von Riechstörungen ärztliche Unterstützung suchen, leidet an einer angeborenen Riechstörung, während erworbene Riechstörungen vergleichsweise seltener auftreten. Die genaue Prävalenz von Riechstörungen im Kindes- und Jugendalter bleibt bisher unklar. Im Vergleich zu Erwachsenen sind Riechstörungen in dieser Altersgruppe jedoch deutlich weniger häufig. Eine grobe Schätzung geht davon aus, dass etwa 1 von 8000 Neugeborenen ohne ausgeprägten Geruchssinn geboren wird.
Diagnostik von Riechstörungen
Um die Riechstörung in ihrer Quantität, Qualität und möglichen Prognose zu erfassen, gilt es eine strukturierte Abklärung der Symptomatik durchzuführen. Die ambulante Basisdiagnostik setzte sich zusammen aus einer ausführlichen Anamnese und einer psychophysische Riechtestung, welche dann je nach Verdachtsdiagnose durch eine spezifische weiterführende Diagnostik ergänzt werden sollte.
Bei der Erstvorstellung sollte eine strukturierte Anamnese erfolgen, die potentielle auslösende Ereignisse, begleitende Symptome, Familienanamnese, relevante Vorerkrankungen und Operationen im HNO Bereich, Medikamenteneinnahmen und etwaige Noxen, berücksichtig. Auch sollte die Betroffenen befragt werden, ob sie an Phantosmien (Gerüche, die nicht vorhanden sind, werden wahrgenommen, z.B. ein geruchloser Raum riecht beißend nach Rauch ohne Geruchsquelle) oder Parosmien (Gerüche werden anders wahrgenommen als sie tatsächlich riechen, z.B. Kaffee riecht nach Hundefutter) leiden. Eine spezifische Familienanamnese hinsichtlich intrafamiliärer Häufung von Riechstörungen erweitert die Anamnese hinsichtlich der Verdachts auf eine angeborene Riechstörung.
Zu beachten ist, dass die alleinige subjektive Einschätzung der Riechfunktion nicht ausreichend ist. Es muss daher eine standardisierte Testung der olfaktorischen Funktion erfolgen.

Im deutschsprachigen Raum erfolgt anschließend die Psychophysische Riechtestung mittels Sniffin‘ Sticks Test. Dieser ist hier am weitesten verbreitet und mit Normdaten im Kindesalter hinterlegt.

Im Kindesalter setzt sich die Testung dabei aus zwei Parameter zusammen. Getestet werden die Geruchsschwelle, bei der die individuelle Wahrnehmungsgrenze eines Geruchs festgestellt wird, und die Geruchsidentifikation, bei der kindgerechte Gerüche mittels Bildkarten identifiziert werden sollen.
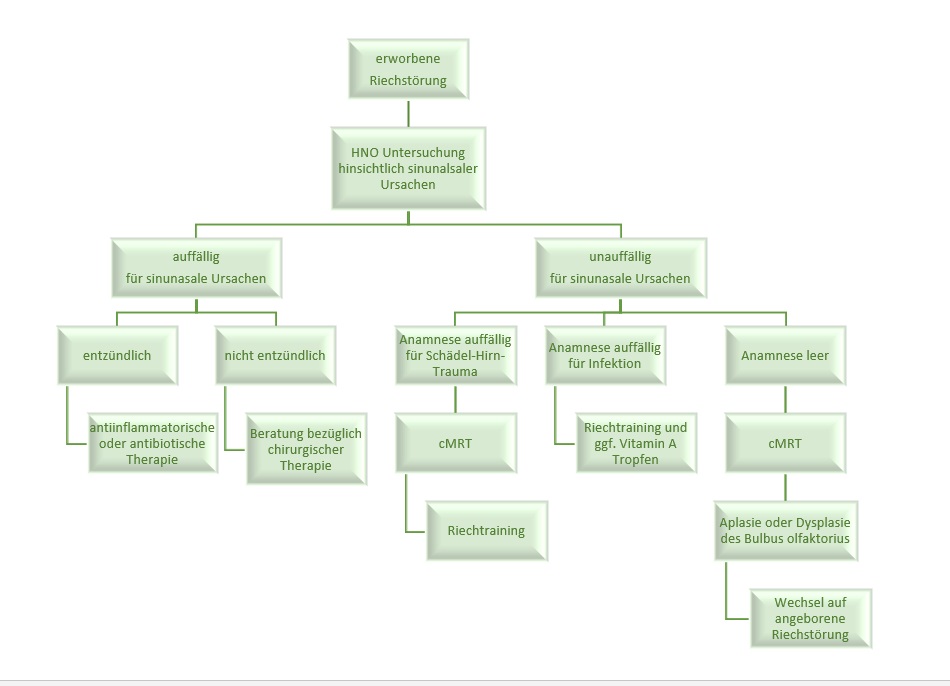
Erworbene Riechstörungen
Zum Ausschluss sinunasaler Riechstörungen im Rahmen der Diagnostik des erworbenen Riechverlustets empfiehlt sich die Überweisung des Betroffenen an eine fachärztliche Kolleg*in der HNO zur Beurteilung möglicher Ursachen wie Polypen oder andere obstruktive Veränderungen der Riechspalte. Sofern keine sinunasale Ursache für die Riechstörung vorliegt, sollten anamnestisch post-infektiöse oder post-traumatische Riechstörungen differentialdiagnostisch abgegrenzt werden.
Um mögliche sekundäre Traumafolgen auszuschließen, sollte bei einem Schädelhirntrauma mit Riechverlust zeitnah eine Schädel-MRT durchgeführt werden. Hierbei ist es ratsam, den Bulbus olfaktorius in die MRT-Untersuchung einzubeziehen, insbesondere mit T2-Wichtung in coronarer Schnittführung und 2 mm Schichtdicke. Nach Ausschluss sinunasaler, post-infektiöser und post-traumatischer Genese, spricht man von einer idiopathischen Riechstörung. Anomalien der anatomischen Strukturen in der Schädel-MRT, wie ein aplastischer oder dysplastischer Bulbus olfactorius, sollten eine erweiterte Diagnostik hinsichtlich einer möglichen kongenitalen Riechstörung nach sich ziehen.
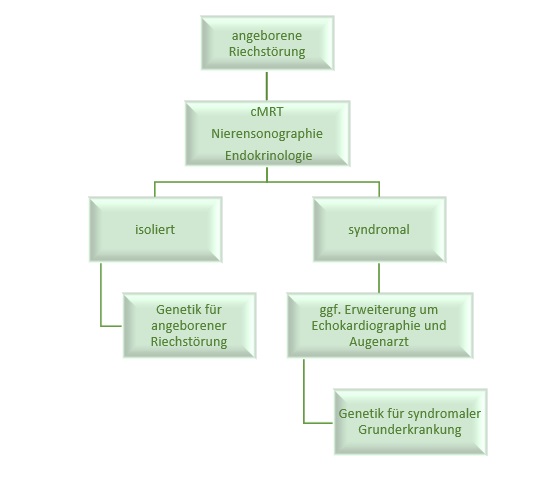
Angeborene Riechstörung
Die Gruppe der angeborenen Riechstörungen unterteilt sich in syndromale Riechstörungen und isolierte kongenitale Anosmien. Zur präzisen Unterscheidung ist es ratsam, eine Umgebungsdiagnostik im Hinblick auf begleitende Fehlbildungen und Fehlfunktionen durchzuführen, die häufig mit Riechstörungen assoziiert sind. Hierzu zählen unter anderem Nierenfehlbildungen und endokrinologische Erkrankungen. Die Diagnostik bei diesen Patient*innen sollte daher eine Schädel-MRT, eine Nierensonographie und eine endokrinologische Untersuchung einschließen. Wenn keine weiteren Auffälligkeiten nachgewiesen werden, besteht ein dringender Verdacht auf eine isolierte kongenitale Anosmie. Auch für diese Erkrankungsform wurden Varianten in verschiedenen Genen (z.B. CNGA2, TENM1, KCNA3 und OBPIIa) beschrieben, daher sollte eine genetische Untersuchung der betroffenen Kinder dringend erwogen werden.